Rowan - Next-Generation Quantum Chemistry
What is Rowan?
While quantum chemical calculations (like density-functional theory and Hartree–Fock) are far superior to forcefields for tasks like geometry optimization, conformer ranking, reaction modeling, and property prediction, quantum chemistry can also be very difficult for non-expert users.
The calculations are very expensive, typically necessitating the use of an external high-performance computing cluster, and interfacing with the requisite software packages typically requires the use of arcane file formats and third-party visualization tools. This complexity deters users from adjacent fields (e.g. drug design, materials science, organic chemistry) who could benefit from the insights that quantum chemistry offers!
Rowan is a web platform that aims to address these issues and make quantum chemistry accessible to non-experts.
How does Rowan work?
Users can upload data in lots of common file formats (sdf, mol2, xyz, mae, SMILES), configure their calculations through a simple GUI, and submit their jobs with the click of a button. Rowan automatically handles allocation of computing resources, job execution, error handling, and real-time display of the results.
The platform operates on a pay-as-you-go model, charging only for the computer time used. Rowan also contains Google Drive-like functionality for organization, annotation, and sharing of calculations.
A particularly helpful feature is the incorporation of the latest AIMNet2 machine-learned interatomic potential from Olexandr Isayev’s group at Carnegie Mellon. AIMNet2 significantly speeds up calculations on drug-like molecules, running several orders of magnitude faster than traditional DFT while maintaining comparable accuracy.
To illustrate the significant time-saving benefits, consider the example of a geometry optimization on apixaban. Utilizing traditional DFT, such a calculation could take hours; however, with the advanced AIMNet2, the same process is completed in a mere two minutes.
For a firsthand look at Rowan in action and to experience the impressive speed enhancements, you can explore the interactive panel below or follow this link.
Optimize multiple structures
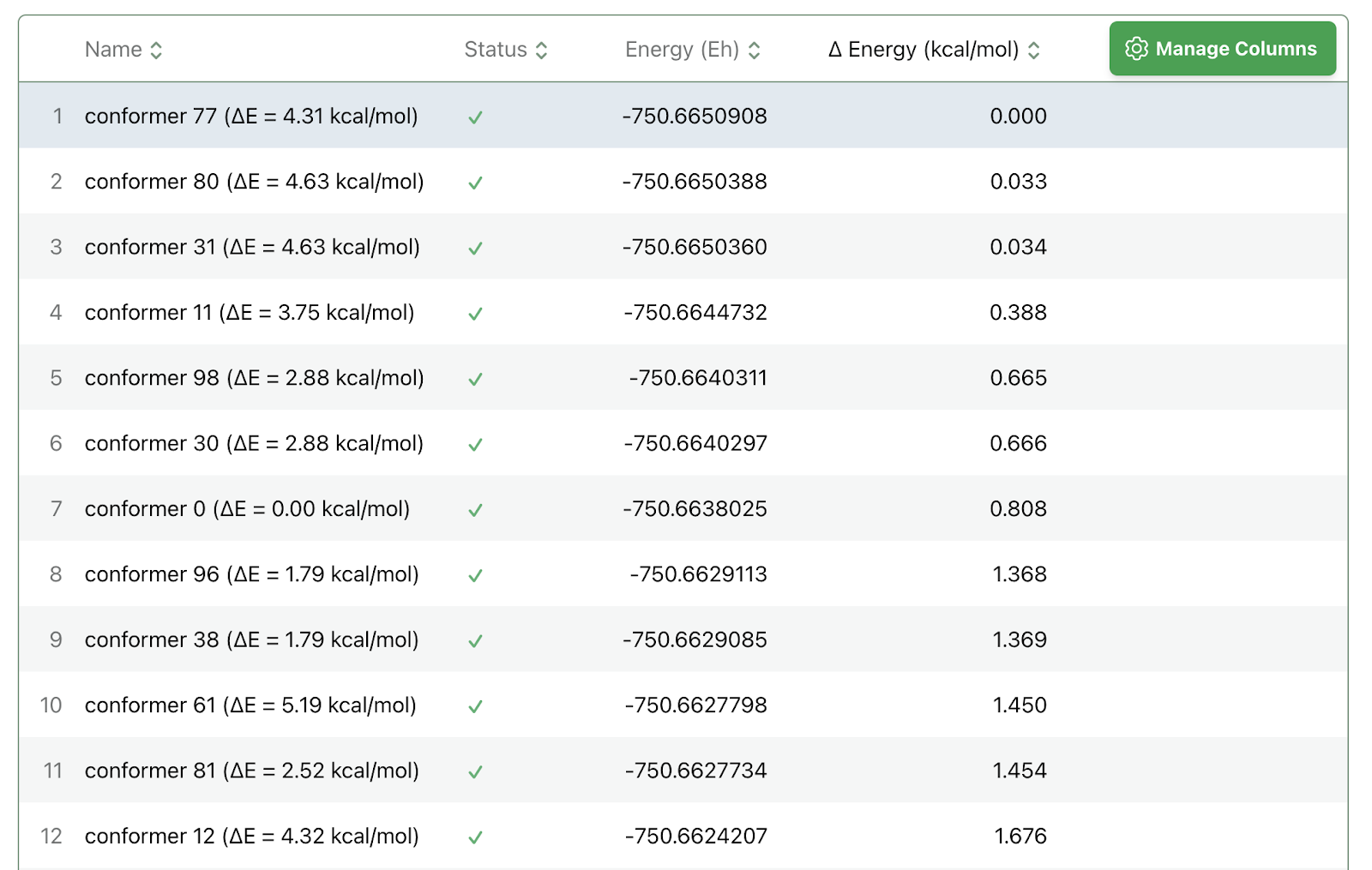
Another handy feature is the “Ensembles” interface, which enables simultaneous submission and analysis of calculations on multiple structures: Rowan can automatically allocate cloud computing resources to run each job in parallel, so ensembles don’t take much longer than a single structure would.
For instance, 70 different conformers of methylphenidate can all be optimized and scored with AIMNet2 in about 4 minutes: the entire ensemble costs 88 credits, or about $1.70. Visualization of the output data demonstrates why reoptimizing conformers with quantum chemistry is so important: the MMFF rankings are incredibly inaccurate!
How to register?
Rowan is still very new, having just launched a few months ago, but already has the potential to be a compelling alternative to conventional programs for plenty of workflows: conformer optimization, dihedral scans, barrier height prediction, and more.
The interface is simple enough to use without any previous experience with quantum chemistry, and you get 500 free credits when you sign up, so it’s worth checking out!
You can make an account with Rowan here.