PyMOL Selection Tool: How to Select Atoms, Residues and Chains (with Examples)
How to select atoms and residues in PyMOL
The PyMOL selection tool lets you isolate any subset of atoms — a single residue, an entire chain, all atoms within 5 Å of a ligand — and then color, show, hide, or analyze them independently. This article is a practical reference: every selection keyword with working syntax and copy-paste examples, covering both the GUI and the select command.
There are two ways to select atoms in PyMOL:
-
The graphical user interface (GUI) — click directly on the structure or use the sequence viewer.
-
The
selectcommand — filter atoms by residue, chain, property, or distance from the command line.
How to Select Atoms in the PyMOL Viewer (GUI)
The first way to select a specific atom or group of atoms is by physically clicking on them in the viewer window. Once you do that, you will notice that:
- The selected residue will be highlighted with pink boxes
- A new object (sele) will appear in the object menu panel.
- In the command line panel you will be prompted with some information such as the chain and residue number of the selected residue.
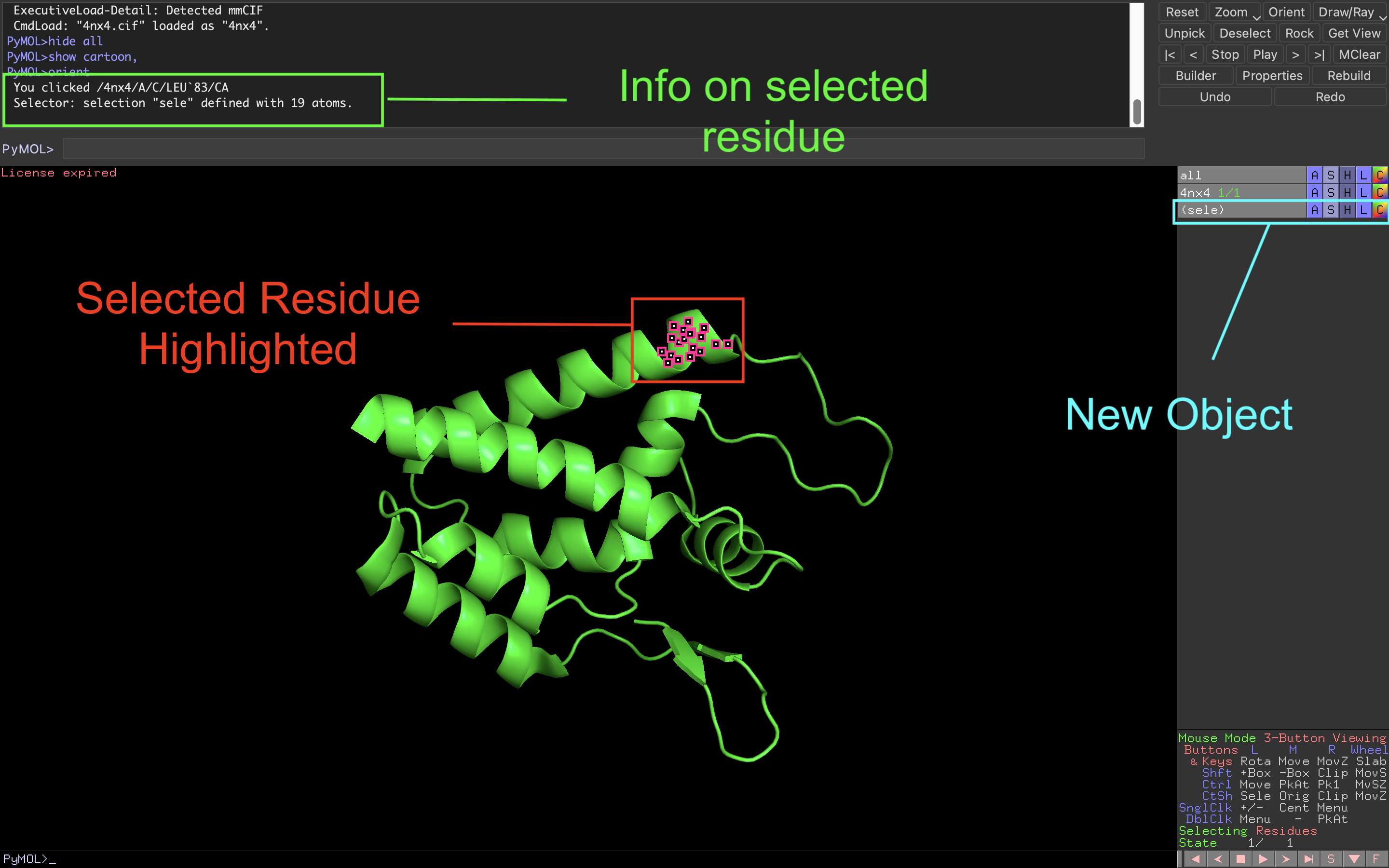
From here, you can grow your selection by clicking on additional residues while you can unselect by clicking on it a second time. To clear all selections, click on an area of the viewer window that does not have any atoms.
To avoid confusion and remember which atoms a selection contains, you can rename it by clicking on A $\rightarrow$ rename selection $\rightarrow$ Type the name and press enter
Then you can use the five pop-up menus available for the selection to modify the properties of the atoms within the group.
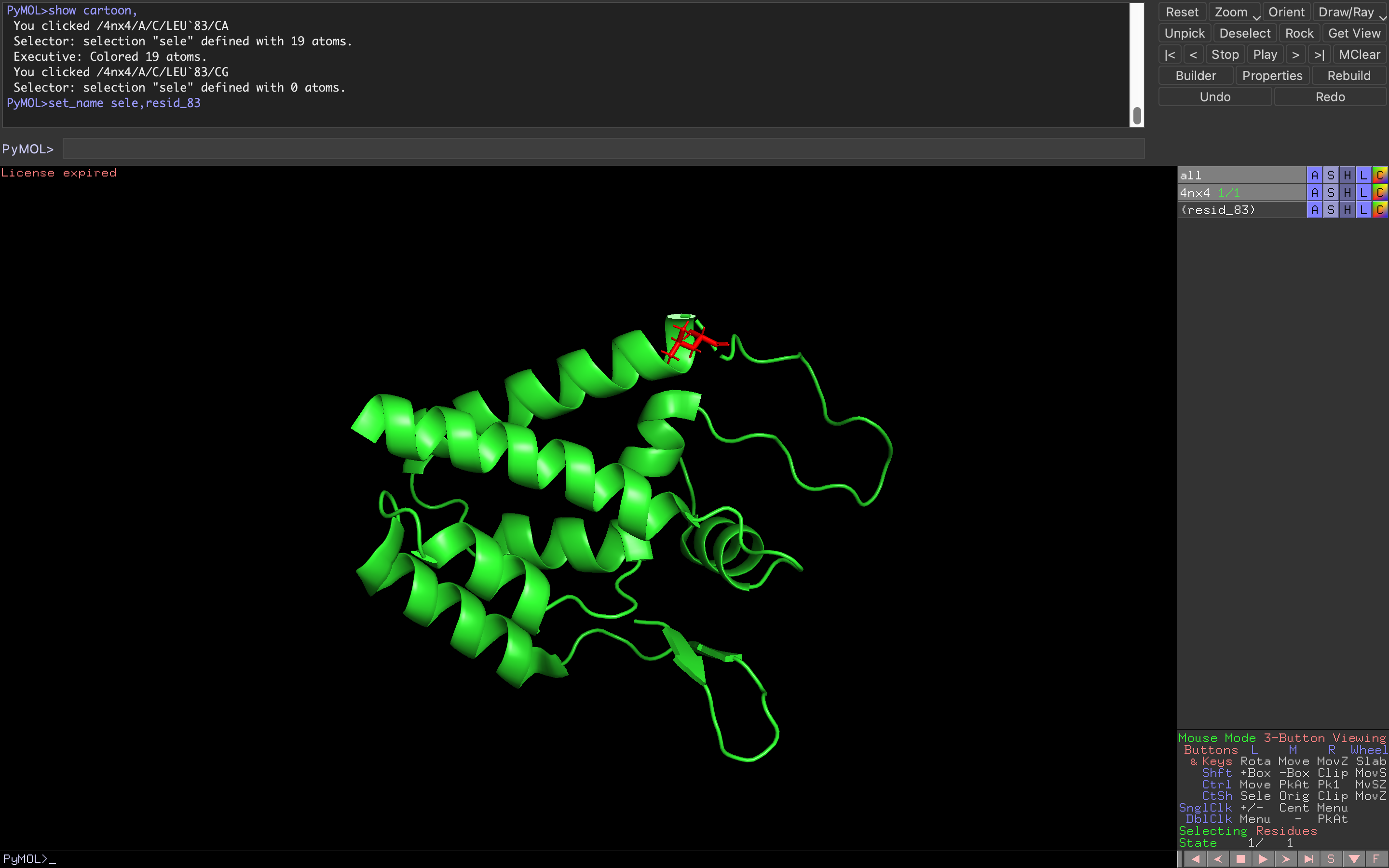
Let’s say that I wanted to display the residue number 83 selected in the previous image to red colored sticks.
- First of all, change the name of the representation to resid_83: A $\rightarrow$ rename selection $\rightarrow$ resid_83
- To change the representation for the selection to sticks: S $\rightarrow$ as $\rightarrow$ sticks
- Finally, change the color of the selected residues to red: C $\rightarrow$ reds $\rightarrow$ red
By default, PyMOL will select residues. Note that you can modify the selection mode by left-clicking on the Selecting mode in the bottom right panel. This will allow you to switch to chain or atom selection among others.
You can also change the selection mode by dragging your cursor up to Mouse $\rightarrow$ Selection Mode $\rightarrow$ choose the selection mode
Selecting by Residue Number Using the Sequence Viewer
The previous approach is extremely convenient when you want to quickly select a residue during a visual inspection but it also comes with some problems. Each time you want to select a residue you first need to find it in the structure.
As you may imagine, it can be quite challenging to interpret complicated structures and find the residue you need. So what if you want to select a specific residue number and you don’t know how to locate it in the protein?
A more practical alternative in such cases is to directly select the residues you need by using the sequence display feature in the GUI window.
This will show the sequence of residues in the protein starting at the N-terminus and ending at the C-terminus. You can then use the scroll bar and click on the residues to select them by number, even if you are not sure of their location in the structure.
To turn on the sequence viewer in PyMOL, you can either click the “S” button below the mouse mode table or navigate to the upper control window and click on Display $\rightarrow$ Sequence.
How to Use the select Command in PyMOL
Selecting regions of your protein by clicking on residues or sequence display is an approach that can be useful when you want to quickly select atoms of interest. However, sometimes you may want to achieve more control over the selection of specific atoms by filtering them according to a series of criteria.
If that is the case, you can use the built-in selection command implemented in PyMOL.
The select command is a powerful and flexible feature of PyMOL that allows users to define complex selections based on a wide range of criteria, including atomic properties, spatial relationships, and chemical properties.
Selection criteria can be refined or broadened by using a selection algebra that combines specific keywords with logical operators (“and”, “or”, not).
The general syntax for a selection in PyMOL is quite intuitive. You only need to call the select command followed by the rules specifying the selection criteria (selection_rule).
|
|
For instance, to select the whole system you can just prompt the select command followed by the specific keyword needed to select everything (all).
|
|
This will create a new selection containing the overall system and having the default name ((sele)) that you can use to perform your analysis.
If you want to create a selection with a custom name you can simply call the command followed by the name you want, and then input the selection rules after the comma:
|
|
In this way, you will generate a selection named my_selection containing all the atoms.
PyMOL Selection Command Reference
The selection algebra in PyMOL is very powerful and allows you to get very creative with your selection operations.
It would be impossible to go through each one of them so here I will give you a few examples of the most useful ones. More specifically, I will focus on three categories of selections:
- Selection by name
- Selection by proximity
- Selection by properties
I will give some examples for each one of them and finally, I will also explain to you how you can combine them using logical operators to create more complex selections.
You can find more info about the selection algebra in the PyMOL wiki.
Select by Residue Name, Number, Atom Name, or Chain
The first few examples we are going to see are selections by name. PyMOL gives you different options to select according to different identifiers (e.g., atom name and number, residue name and number, chain, …) as specified in the PDB file or any other file format you are using.
Atom name selection:
If you want to select by atom name you can do that by calling the select command followed by the name keyword and the atom name (<atom_name>).
|
|
Let’s say you want to group all the $C_{\alpha}$ in your protein in a selection named CA :
|
|
You can also create a selection with different atom names such as $C_{\alpha}$, and $C_{\beta}$:
|
|
Atom number selection:
To select by atom number we need to use the id identifier.
|
|
You can select a certain atom number (e.g., 10) as well as a range of atoms (1-100):
|
|
Residue name selection:
To select by residue name use the resn keyword followed by the name of the residue you want to select (<residue_name>).
|
|
You can select all the Alanines in your protein in a selection named ALA :
|
|
And add Glycine to the selection using the + sign:
|
|
Residue number selection:
To select by residue number we need to use the resi identifier.
|
|
You can select a certain residue number (e.g., 10) or a range of residues (1-100):
|
|
Chain identifier selection:
Some proteins may be arranged in different chains. If that is the case, PyMOL also gives you the possibility to select by chain using the chain keyword followed by the chain identifier (<chain_id>).
|
|
To select chain A of your protein:
|
|
Select by Distance: within and bound_to
Another possible option is to select by proximity, namely depending on the surrounding environment of a given atom. Here we will consider two use cases.
1. The first one will be the within keyword that allows you to select atoms in a selection (<sele_1>) that are within a certain distance (<distance> in Å) of another atom or group of atoms (<sele_2>):
|
|
This statement allows you to select all the $C_{\alpha}$ within 10 Å of a residue range (1-10) and includes them in a selection name CAs.
|
|
2. The second example will discuss the bound_to keyword that lets you select all the atoms bounded to a certain selection (<my_selection>).
|
|
The following command allows you to select all the atoms bounded to residue number 70 and includes them in a selection name bounded_70.
|
|
Select by Properties: Secondary Structure, B-Factor, and Chemical Class
PyMOL also gives you the possibility to select atoms depending on a different set of properties of atoms. Here we will briefly consider some of them including secondary structure, b-factor, and chemical class.
Select by secondary structure
To select by secondary structure you can use the ss keyword and then specify the secondary structure you want to select (<ss>).
|
|
The values of <ss> can be either h = helix or s = sheet. You can also select both of them by adding the + sign.
|
|
Select by b-factor
The b-factor is an important parameter indicating the most mobile regions of a protein. If you want to select atoms based on their b-factor, you can use the b keyword followed by the "<", “>”, “=” operators, and a cutoff value (<cutoff>):
|
|
To include all the atoms having b-factor less than 10:
|
|
Select by chemical class
PyMOL also gives you a series of specific keywords that you can use to group atoms based on the chemical class they belong to. Some of them are reported in the table.
| Keyword | Class | Command |
|---|---|---|
organic |
organic compounds (e.g., ligands) | select, organic |
solvent |
water molecules | select, solvent |
hydrogens |
hydrogen molecules | select, hydrogens |
backbone |
backbone atoms | select, backbone |
sidechain |
sidechain atoms | select, sidechain |
metals |
metal atoms | select, metals |
Combining Selections with and, or, and not
The main advantage of the select command is that you can create complex selection expressions by combining all of the rules that I previously showed you using logical operators. The three logical operators available in PyMOL are and, or, and not.
Here are some examples of how you can use these operators to improve the selection in PyMOL:
1. The and operator is used to specify that the selection should include only atoms that meet both of the specified criteria.
Let’s say that you want to group all the $C_{\alpha}$ in the residue range 50-60 protein in a selection named CA_50_60. You can use the and operator to combine the keyword selecting the residue range (resi 50-60) and the one selecting the $C_{\alpha}$ (name CA).
|
|
2. Use the or to specify that at least one of multiple selection criteria must be met.
Select every residue in the range of 10-20 or 30-40 in your protein. You can use the or operator to associate the keywords to select both of the residue ranges (resi 10-20, resi 30-40)
|
|
3. The not logical operator reverses the expression that immediately follows.
For instance, if you want to select everything except for $C_{\alpha}$ you can use the keyword needed to select the alpha carbons (name CA) preceded by the not operator.
|
|
You can also create a selection with multiple logical operators at the same time.
To select every $C_{\alpha}$ in chain A or every $C_{\beta}$ in chain B. Just make sure to use parenthesis in the appropriate way to avoid confusion and that the operations are performed in the right order.
|
|
How to Apply a Selection: show, hide, and color
Once you have selected the atoms of interest you can use them to perform operations, such as displaying, hiding, changing their representation, or coloring them. Alternatively, certain commands allow you to directly perform a selection operation after the comma.
1. Visualize the selection: you can use the show, and hide commands to display or hide the selected atoms in the PyMOL viewer (my_sele) using your favorite representation. For example:
|
|
2. Change the color of the selection: You can use the color command to change the color of the selection.
|
|
Find more info on the coloring and customization process in PyMOL here