RMSD in GROMACS: what it measures and how to calculate it with gmx rms
The gmx rms command
RMSD (Root Mean Square Deviation) is one of the first things you check after an MD simulation. It tells you how much the structure drifted from the starting geometry and whether the system has reached equilibrium. This article explains what RMSD measures, derives the equation, and shows you how to calculate it step by step with gmx rms in GROMACS.
What Does RMSD Measure in MD Simulations?
The Root Mean Square Deviation (RMSD) measures how much a certain molecular structure deviates from a reference geometry.
This feature plays a key role in the field of molecular modeling, and you will find it referenced in various papers.
But why is it so important?
In Molecular Dynamics (MD), we are interested in how our system evolves over time as compared to its starting structure. For this reason, plotting the RMSD as a function of time can help us in two ways:
-
First of all, it is useful to identify parts of the simulation where we have important changes in the structure. For example, if a protein over the course of the simulation has some large conformational changes we should notice a spike in the RMSD vs. time plot.
-
As a second point, The RMSD is commonly used as an indicator of convergence of the structure towards an equilibrium state. A flattening of the RMSD curve can indicate that the protein has equilibrated and that we can proceed with the next step.
Therefore, the RMSD has a critical role in the different steps of an MD simulation (i.e., equilibration and production runs).
The equation for RMSD
Let’s discuss the formula needed to compute this fundamental structural feature.
For a general molecular structure with $N$ atoms, we can compute the RMSD between a certain conformation ($r$) and a reference structure ($r^{ref}$) via the following equation:
$$ RMSD = \sqrt{\frac{1}{N}\displaystyle\sum_{i=1}^N(r_i - r_i^{ref})^2} $$
where $r_i^{ref}$ represents the set of coordinates of the $i$-th atom in the reference structure, while $r_i$ is the set of coordinates of the same atom but belonging to the structure that is going to be compared to the reference.
$$ r_i = x_i + y_i + z_i $$ $$ r_i^{ref} = x_i^{ref} + y_i^{ref} + z_i^{ref} $$
In summary, the RMSD is the square root of the average of the squared distances between atoms.
During a simulation, we actually compute the RMSD as a function of time $t$.
At each time step, we compute the RMSD between the coordinates at that specific time step $r(t)$, and the reference.
$$ RMSD(t) = \sqrt{\frac{1}{N}\displaystyle\sum_{i=1}^N(r_i(t) - r_i^{ref})^2} $$
In this way, we can see how the system deviates from the reference structure during our MD simulation.
How the gmx rms Command Works
In GROMACS we compute the RMSD via the gmx rms command.
The workflow for this command is quite intuitive. You only need two things:
- A reference geometry in the tpr or gro format
- A trajectory file that is going to be compared to the reference (xtc file)
GROMACS will output a file in the xvg format that you can plot using Grace or Python as I showed in this article .
Here I am going to show you a few examples of how you can play with the gmx rms command to compute the RMSD of your system.
Putting this into practice? RMSD is usually the first thing you check once a simulation finishes. My complete GROMACS protein-in-water guide takes you all the way there, from a PDB file to a trajectory you can run gmx rms on, with every MDP file and script included. Get the guide →
Calculate RMSD Along a Full Trajectory
First of all, we are going to consider the most basic example.
How can we use the gmx rms command to measure the RMSD fluctuation of the system along the simulation?
In other words, we want to observe how much the structure of the system deviates from the starting one.
In GROMACS this is accomplished by comparing each frame in the trajectory file with the reference structure.
To this aim, you can type the following:
|
|
- Call the program with
gmx - Select the
rmscommand - Select the
-fflag and provide the xtc file from your simulation (simulation.xtc) - Choose the reference structure (
reference.tpr) with the-sflag - Call the
-oflag and decide how you want to name the output xvg file (rmsd.xvg)
- You may want to change the time unit with the
-tu nsoption to output the xvg file in terms of ns instead of ps.
At this stage, you will be prompted to select two groups.
1. The group for least square fits: structure under -f flag
|
|
2. The group for RMSD calculation: structure under -s flag
|
|
For protein simulations, it is a common practice to compute the “backbone to backbone” RMSD value (Select group “4” for both).
You can automate the selection with this command:
|
|
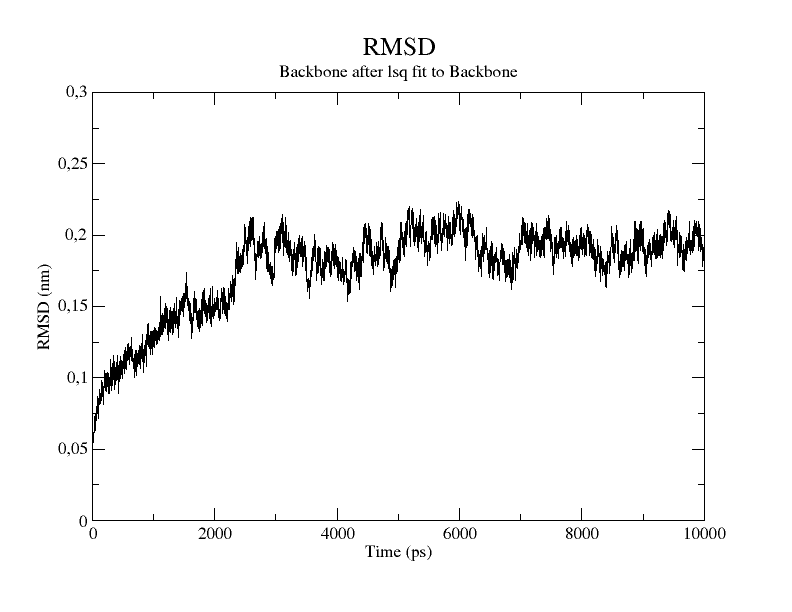
The resulting rmsd.xvg file can be plotted with Grace.
|
|
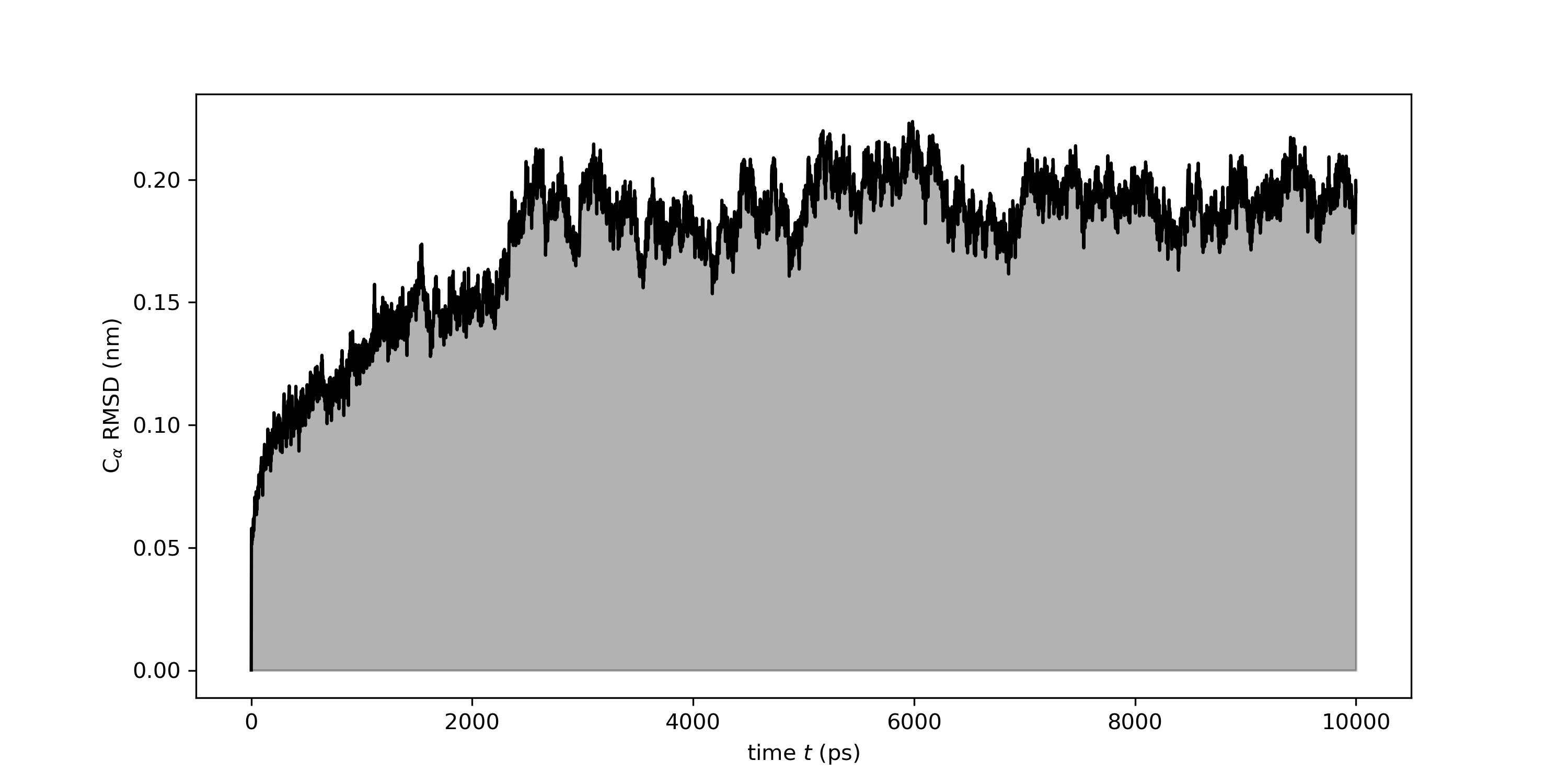
Or with Python using the Numpy and Matplotlib libraries.
|
|
How to Interpret the RMSD Plot
Once you have plotted rmsd.xvg, here is what the curve shape tells you.
A plateau: the RMSD rises initially then levels off and stays flat. This is the expected pattern. The rising phase is the system relaxing from the starting structure; the plateau indicates the protein has equilibrated and is sampling a stable basin.
A continuous drift: the RMSD keeps climbing throughout the simulation without flattening. The simulation may be too short, or the starting structure was far from equilibrium.
Sudden spikes: a sharp jump followed by a drop back to the previous level usually indicates a transient conformational event which might need to be analyzed further,
For a small globular protein, backbone RMSD plateau values in the range of 0.1–0.2 nm are typical, but larger values are not necessarily wrong, as it depends on the size and flexibility of your system. There is no universal threshold. What matters is the overall shape: if the curve looks obviously wrong (extreme values, persistent spikes, never converges), the first thing to do is load the trajectory in VMD and visually inspect what is happening to the protein. A common problem might be due to PBC artifacts.
Calculate RMSD for a Specific Group Using an Index File
If you want to analyze a specific part of your system that is not within the default GROMACS groups you can create an index with the gmx make_ndx command.
Subsequently, you can pass the special index that you created to the gmx rms command.
|
|
- Call the program with
gmx - Select the
rmscommand - Select the
-fflag and provide the xtc file from your simulation (simulation.xtc) - Choose the reference structure (
reference.tpr) with the-sflag - Call the
-oflag and decide how you want to name the output xvg file (rmsd_index.xvg) - Prompt the special index you created (
index.ndx) via the-nflag
Calculate RMSD Over a Specific Time Window
You can compute the RMSD for a specific time frame of your trajectory by using the -b and -e flags.
For instance, if you are only interested in measuring the RMSD between 1000 ps and 2000 ps of your trajectory you can use the following command.
|
|
- Call the program with
gmx - Select the
rmscommand - Select the
-fflag and provide the xtc file from your simulation (simulation.xtc) - Choose the reference structure (
reference.tpr) with the-sflag - Call the
-oflag and decide how you want to name the output xvg file for that specific time frame (rmsd_time.xvg) - Pick the starting time (1000 ps) for the RMSD calculation using the
-bflag - Select the ending time (2000 ps) using the
-eoption
By selecting the same value for the -b and -e flags you will get the RMSD value between the reference and the frame corresponding to the time you selected.